Mucolipidosis alpha/beta
Mucolipidosis alpha/beta
Mucolipidosis alpha/beta can be categorised into Mucolipidosis II, II/III or III, depending on the severity of the symptoms.
Mucolipidosis II alpha/beta (I-Cell Disease) and Mucolipidosis III alpha/beta (Pseudo-Hurler Polydystrophy) were first described in the 1960s. The more severe of the two, I-Cell Disease, was named by Jules Leroy, a Belgian paediatrician and geneticist, for the inclusions he saw in effected individuals’ cells under a microscope. ML III (Pseudo-Hurler Polydystrophy) was described by the French physicians, Maroteaux and Lamy, who found the disease reminiscent of severe MPS I, though milder in its manifestations. Both diseases were assigned to the class of Mucolipidosis, I-Cell Disease as Mucolipidosis II and Pseudo-Hurler Polydystrophy as Mucolipidosis III (or simply, ML II and III). Recent molecular studies identified mutations that correlate with the phenotypes (characteristics) seen in the spectrum of Mucolipidosis II and III, leading to revised classification.

Revised Classification of Mucolipidosis II and III
| Past Nomenclature | Current Nomenclature | |
| I-Cell Disease | ML II | ML II alpha/beta |
| Pseudo-Hurler Polydystrophy | ML IIIA | ML III alpha/beta |
| ML III variant | ML IIIC | ML III gamma |
Gene Mutations and the link between ML II, ML II/III and ML III
ML II and ML III are related conditions on the same spectrum of severity. Most individuals have definite features of ML II, the more severe disease, or definite features of ML III, the milder disease. Intermediate ML II/III is the descriptive name used for a subset of individuals who show small stature and skeletal findings similar to individuals with ML II but better cognitive function and longer lifespan, similar to individuals with ML III. Knowing the gene changes in GNPTAB further helps classify the severity of illness. People with ML II have two significant mutations, which thoroughly impair the normal activity of the phosphotransferase enzyme. People with ML III have one or two milder mutations, leading to a small amount of normal enzyme activity. People with Intermediate ML II/III have specific gene mutations that lead to the intermediate clinical features. For this reason, knowing the gene changes can lead to a more accurate expectation of prognosis in an affected individual. The following report presents and discusses the clinical and biochemical findings of the ‘intermediate’ ML in eight patients with the c.10A4C missense mutation in exon 1 in their compound heterozygous GNPTAB genotype: A novel intermediate mucolipidosis II/III alpha/beta caused by GNPTAB mutation in the cystosolic N-terminal domain
ML II alpha/beta (I-Cell Disease)
Mucolipidosis II alpha/beta (also known as I-Cell Disease) is a progressively debilitating disorder that affects many parts of the body and is caused by mutations in the GNPTAB gene. This gene provides instructions for making part of an enzyme called GlcNAc-1-phosphotransferase. Mutations in the GNPTAB gene that cause mucolipidosis II alpha/beta prevent the production of any functional GlcNAc-1-phosphotransferase
At birth, children with mucolipidosis II alpha/beta are small and have weak muscle tone (hypotonia) and a weak cry. Affected individuals grow slowly after birth and usually stop growing during the second year of life. Development is delayed, particularly the development of speech and motor skills such as sitting and standing.
Children with mucolipidosis II alpha/beta typically have several bone abnormalities, many of which are present at birth. Affected individuals may have an abnormally rounded upper back (kyphosis), feet that are abnormally rotated (clubfeet), dislocated hips, unusually shaped long bones, and short hands and fingers. People with this condition also have joint deformities (contractures) that significantly affect mobility. Most children with mucolipidosis II alpha/beta do not develop the ability to walk independently. Affected individuals have dysostosis multiplex, which refers to multiple skeletal abnormalities seen on x-ray.
Other features of mucolipidosis II alpha/beta include a soft out-pouching around the belly-button (umbilical hernia) or lower abdomen (inguinal hernia), heart valve abnormalities, distinctive-looking facial features that are described as “coarse,” and overgrowth of the gums (gingival hypertrophy). Vocal cords can stiffen, resulting in a hoarse voice. The airway is narrow, which can contribute to prolonged or recurrent respiratory infections. Affected individuals may also have recurrent ear infections, which can lead to hearing loss.
The A to Z of Mucolipidosis II
Anaesthetics and Airway
Dr Leroy and Dr Sara Cathey from the Greenwood Genetics Centre have very kindly written a Medical Alert for families whose children may need an anaesthetic. This document can be viewed on the website or downloaded as a PDF . It is critically important to know that children with ML II have very small and stiff airways. Advanced intubation techniques are required. Removing the breathing tube (extubation) can be equally complicated in some individuals. Any elective surgery, even routine or simple procedures, should take place in a tertiary care center equipped to handle complicated patients with rare diseases.
Abdomen and Hernias
A child with ML II is likely to have a protruding abdomen due to shortness of the chest, posture, or weakness of the muscles. Significant enlargement of liver and spleen is less common in ML II or III than in other lysosomal diseases. The liver and the spleen may be felt (palpated) by a doctor who examines your child because of this combination of factors.
Frequently, part of the abdominal contents will push out behind a weak spot in the wall of the abdomen. This is called a hernia. The hernia can come from behind the navel (umbilical hernia) or in the groin (inguinal hernia). Inguinal hernias should be repaired by an operation, but hernias sometimes recur. Umbilical hernias are not usually treated unless they cause entrapment of the intestine.
Bones and Joints
Joint stiffness is common in both ML II and III and the maximum range of movement of all joints may become limited.
There is variation in the severity of problems that affect bones and joints, even between affected brothers and sisters. Those with ML II are affected early and skeletal problems may even be evident at birth. Anti-inflammatory drugs, such as ibuprofen, can help with joint pain, but their use should be monitored closely to make sure that irritation and ulcers in the stomach do not occur. Some individuals with ML II have decreased bone density leading to osteopenia and osteoporosis, also called secondary metabolic bone disease. Bisphosphonates have been trialled in a good number of ML II and ML III patients reducing pain, maintaining mobility and improving quality of life. Your orthopedist or endocrinologist can provide additional information about this treatment.
Spine
The bones of the spine (vertebrae) normally line up from the neck to the buttocks.
Individuals with ML II often have poorly formed vertebrae that may not stably support each other. One or two of the vertebrae in the middle of the back are sometimes slightly smaller than the rest and set back in line. This backward slippage of the vertebrae can cause an angular curve (kyphosis or gibbus) to develop, but it usually does not need treatment, unless severe.
Neck
Rarely, the bones that stabilize the connection between head and neck may be malformed (odontoid dysplasia) in people with ML II. If this occurs, fusion surgery may be required to connect all bones to each other so they do not slip further.
Parents of children with ML II should be cautious about how the area of the spine around the neck is handled. Activites that alter the stability of the neck (such as trampolining) should be avoided.
Hands
The shape of the hands is very noticeable in ML II; the hands are short and broad with stubby fingers. The fingers stiffen and gradually become curved, due to limited joint movement. The tips of the fingers can become permanently bent over.
Breathing Difficulties
 Many affected individuals breathe very noisily even when there is no infection. At night they may be restless and snore. Sometimes the individual may stop breathing for short periods while asleep (sleep apnea). Pauses of up to 10-15 seconds may be considered normal. This noisy breathing, which stops and starts, can be very frightening for parents to hear. They may fear that their child is dying. If this is happening, the child’s oxygen level may be low when sleeping which can cause problems with the heart. If a parent notices significant choking or episodes of interrupted breathing, the child should be evaluated by a sleep specialist using a polysomnogram (sleep study). It is important to know that many individuals may breathe like this for years. Sleep apnea can be treated in some individuals by removing the tonsils and adenoids, opening up the airway with night-time CPAP treatment (continuous positive airway pressure), BiPAP (bi-level positive airway pressure), or tracheostomy, as discussed in the following paragraphs. There are reports of tonsils and adenoids growing back and continuing to cause airway problems.
Many affected individuals breathe very noisily even when there is no infection. At night they may be restless and snore. Sometimes the individual may stop breathing for short periods while asleep (sleep apnea). Pauses of up to 10-15 seconds may be considered normal. This noisy breathing, which stops and starts, can be very frightening for parents to hear. They may fear that their child is dying. If this is happening, the child’s oxygen level may be low when sleeping which can cause problems with the heart. If a parent notices significant choking or episodes of interrupted breathing, the child should be evaluated by a sleep specialist using a polysomnogram (sleep study). It is important to know that many individuals may breathe like this for years. Sleep apnea can be treated in some individuals by removing the tonsils and adenoids, opening up the airway with night-time CPAP treatment (continuous positive airway pressure), BiPAP (bi-level positive airway pressure), or tracheostomy, as discussed in the following paragraphs. There are reports of tonsils and adenoids growing back and continuing to cause airway problems.
Management of breathing problems
The doctor may want the child to be admitted to the hospital overnight for a sleep study. Monitors are placed on the skin and connected to a computer to measure the levels of oxygen in the blood, breathing effort, brain waves during sleep and other monitors of the body’s function. From this study, the doctors can assess how much blockage to breathing is present, how much trouble your child is having moving air into the lungs during sleep, and how much effect this has on his body.
Night time CPAP or BiPAP can open the airway at night using air pressure, which can help the child’s airway stay open. This treatment involves placing a mask on the face each night and having air pumped into the airway to keep it from collapsing. This may seem to be an extreme measure but many people are able to tolerate it because it can greatly improve the quality of sleep, as well as help prevent or reduce the risk of heart failure caused by low oxygen levels at night.
Carpel Tunnel Syndrome
Individuals with ML II may experience pain and loss of feeling in the fingertips caused by carpal tunnel syndrome. The wrist or carpus consists of eight small bones known as the carpals, which are joined by fibrous bands of protein called ligaments. Nerves have to pass through the wrists in the space between the carpal bones and the ligaments. Thickening of the ligaments causes pressure on the nerves, and this can cause irreversible nerve damage. The nerve damage will cause the muscle at the base of the thumb to waste away and will make it hard for a child to oppose his thumb in a position for a normal grasp. Although your child may not complain of pain, carpal tunnel syndrome may be severe. If your child seems to have pain in the hands, particularly at night or upon waking, an electrical test called a nerve conduction or electromyograph study should be performed. This test will show whether carpal tunnel syndrome is the cause. If your child has any weakness at all in the hand or has decreased muscle mass at the base of the thumb, then ask for the test from your neurologist. Be persistent, as many physicians may not believe that carpal tunnel syndrome is present without the classic symptoms.
Any elective surgery, even routine or simple procedures, should take place in a tertiary care centre equipped to handle complicated patients with rare diseases.
Uncorrected carpal tunnel syndrome may result in the loss of sensation or the sensation of pins and needles in the hands and fingers. Carpal tunnel syndrome can be corrected through surgery.
A similar type of nerve compression can happen elsewhere in the body, such as the feet, and cause localized weakness or pain.
Development and Intelligence
Children with ML II have significant developmental delays, in both motor skills and language. Few are ever able to walk independently, without support. Spoken language may be limited to few words or phrases but understanding is generally much better. Children with ML II are aware of and interact with their immediate environment. They show happiness and sadness, and have likes and dislikes, just as any child does.
Diet
There is no scientific evidence that a particular diet has any helpful effect on people with ML II. symptoms such as diarrhoea or constipation are as common in ML II as in the general population.
It is important to note that there is no diet that can prevent the storage of compounds normally degraded in lysosomes, because the compounds are actually created by the body. So reducing sugar intake or other dietary components cannot reduce storage.
Some children with ML II are not tolerant of hard food textures, preferring soft foods. This may be the result of the tongue thickening or swallowing complications. If this occurs, an evaluation by a pediatric nutritionist is recommended to ensure the child is receiving adequate nutrition. Many children with ML II have a limited number of foods in their diet, out of preference.
Ears
It is important that individuals with ML II and III have their hearing monitored regularly so that problems can be treated early. Frequent ear infections may contribute to conductive hearing loss.
Chronic middle ear effusion
Correct functioning of the middle ear depends on the pressure behind the eardrum being the same as that in the outer ear canal and the atmosphere. This pressure is equalized by the eustachian tube, which runs to the middle ear from the back of the throat. If the tube is blocked, the pressure behind the eardrum will drop and the drum will be drawn in. If this negative pressure persists, fluid from lining of the middle ear will build up and eventually become thick like glue. This is called middle ear effusion. An ENT physician may advise that grommets/ear tubes be placed if chronic middle ear effusion is causing pain, recurrent infection, or impaired hearing in your child. Remember, any surgical procedure, even simple ear tubes, should take place in a tertiary care centre equipped to handle complicated patients with rare diseases.
Education
Children with ML II may benefit from having a mainstreamed education and enjoy the social interaction with peers. It is important to work with your school system and develop the best Individualized Education Program (IEP) for your child. The IEP should emphasize the child’s mobility limitations, adaptive physical education, and attention to fine motor modifications.
Eyes
 Children with ML II may be affected by mild corneal clouding. The circular window at the front of the eye (cornea) becomes cloudy which disrupts the clear layers of the cornea. Corneal clouding is rarely severe in ML II. Routine eye examinations by an ophthalmologist are recommended.
Children with ML II may be affected by mild corneal clouding. The circular window at the front of the eye (cornea) becomes cloudy which disrupts the clear layers of the cornea. Corneal clouding is rarely severe in ML II. Routine eye examinations by an ophthalmologist are recommended.
Growth
Birth weight and length may be smaller than expected in babies with ML II. Children with ML II stop growing around or before 2 years of age, and they remain small. Usually children with ML II take in the calories they need by mouth. Tube feedings might be needed in some circumstances, but it is important to recognize that extra calories cannot “make” a child with a disease that causes small stature grow taller.
Heart
Heart disease can occur in ML II.
During examinations, your doctor may hear a sound in your child’s heart, called a heart murmur. A murmur may be a normal finding or it may indicate a heart problem. A simple and painless test called an echocardiogram (similar to ultrasound screening of babies in the womb) is used to identify the problem. Heart valves are designed to close tightly to prevent blood from flowing in the wrong direction as blood passes from one chamber of the heart to another. If a valve becomes weakened or hardened, it may not shut firmly enough and a small amount of blood may leak through the valve. It is possible to have heart valve problems for years without any ill effects. If problems do occur, it may be possible to treat them with surgery.
Children with ML II can also develop the more serious problems of thickening and weakness of the heart muscle (cardiomyopathy) and congestive heart disease. Heart medications may be prescribed. Because of the unusual special problems that can occur in these diseases, you should select a cardiologist with some knowledge of Mucolipidosis. At a minimum, you should inform the doctor about the problems experienced by individuals with ML diseases.
Life expectancy
Life expectancy in ML II is shortened, and survival beyond the first decade is not common.
Mouth
Children with ML II generally have overgrown and swollen gums, which are characteristic of the disease. Their teeth may be late and troublesome in breaking through the gum. Teeth may appear just above or below the edge of the gum and are often widely spaced and poorly formed with fragile enamel. Sometimes the tongue may be enlarged, and the roof of the mouth may have a high arch.
It is important that the teeth are well cared for, as tooth decay can be a cause of pain. Teeth should be cleaned regularly. Cleaning inside the mouth with a small sponge on a stick soaked in mouthwash will help keep the mouth fresh and help avoid bad breath. Even with the best dental care, an abscess around a tooth can develop due to abnormal formation of the tooth.
Since individuals with ML may have thickening of the heart valves, you may be advised to give your child prescribed antibiotics before and after any dental treatment. This is because certain bacteria in the mouth may get into the bloodstream and cause an infection in an abnormal heart valve, potentially damaging it further. If teeth need to be removed while under an anaesthetic, it should be done in the hospital under the care of both an experienced anesthetist and a dentist – never in the dentist’s office.
Neurological problems: Brain, Senses and Nerves
Impaired cognitive development is characteristic of ML II. Other aspects of ML II can affect brain function, including inadequate oxygen levels, sleep deprivation due to sleep apnea, increased fluid pressure in and around the brain (hydrocephalus), and effects on the eyes and ears that affect the ability of the individual to see and hear normally.
The brain and the spinal cord are protected from jolting by the cerebrospinal fluid that circulates around them. Uncommonly, in individuals with ML II, the circulation of the fluid may become blocked over time so that it cannot be taken back into the bloodstream. The blockage (communicating hydrocephalus) causes increased pressure inside the head, which can press on the brain and cause headaches, incontinence, delayed development, expansion of the skull, and impaired vision. If hydrocephalus is suspected, an MRI should be performed. However, a lumbar puncture with pressure measurement (ideally pressure monitoring) is the best way to assess if increased intracranial pressure exists.. If your doctor confirms that the individual has elevated intracranial pressure, it can be treated by the insertion of a thin tube (shunt) that drains fluid from the brain into the abdomen (ventriculoperitoneal or VP shunt). The shunt has a pressure sensitive valve that allows spinal fluid to be drained to the abdomen when the pressure around the brain becomes too high. The lack of papilledema (swelling around the optic disk) or normal sized ventricles does not rule out hydrocephalus in individuals with ML II.
Nose, throat chest and ear problems
Runny nose
Typically, the bridge of the nose is flattened and the passage behind the nose may be smaller than usual due to poor growth of the bones in the mid-face and thickening of the mucosal lining. This combination of abnormal bones, with storage in the soft tissues in the nose and throat, leads to a narrow airway which can become easily blocked. Individuals with ML II can have chronic discharge of thick clear mucous from the nose (rhinorrhea), and chronic ear and sinus infections.
Throat
The tonsils and adenoids often become enlarged and can partly block the airway. The neck is usually short, which contributes to problems in breathing. The windpipe (trachea) becomes narrowed by storage material and may be stiff, due to abnormal cartilage rings in the trachea. Individuals with ML II may have hoarse sounding voices due to stiff vocal cords.
Chest
The chest is short and the shape of the chest is frequently abnormal as the junction between the ribs and the breastbone (sternum) is not as flexible as it should be. The chest is therefore rigid and cannot move freely to allow the lungs to take in a large volume of air. The muscle at the base of the chest (diaphragm) is pushed upwards if the liver and spleen are enlarged, further reducing the space for the lungs. When the lungs are not fully cleared, there is an increased risk of infection (pneumonia).
Treatment of respiratory infections
It is essential to consult your doctor before using over-the-counter medications. Drugs for controlling mucous production may not help. Drugs, such as antihistamines, may dry out the mucous, making it thicker and harder to dislodge. Decongestants usually contain stimulants that can raise blood pressure and narrow blood vessels, both undesirable for people with ML II. Cough suppressants or drugs that are too sedating may cause more problems with sleep apnea by depressing muscle tone and respiration.
Although most children with colds do not require antibiotics, individuals with Mucolipidosis frequently contract secondary bacterial infections of the sinuses or middle ear. Bacterial infections should be treated with antibiotics. Poor drainage of the sinuses and middle ear makes overcoming infections difficult. Therefore, it is common to have infections improve on antibiotics and then promptly recur after the antibiotic course is over. Chronic antibiotic therapy might be used to help some individuals with recurring ear infections. While overusing antibiotics is dangerous because of the risk of antibiotic-resistant infections, appropriate use of antibiotics is important in people with ML. You will need a doctor with whom you can develop a good working relationship to manage the frequent infections. Ventilation tubes can be used to improve drainage from the ear and speed resolution of infections. It is important to consult with an Ears, Nose and Throat (ENT) specialist experienced with ML diseases to determine which type of tube is best. Any elective surgery (even simple ear tubes) should take place in a tertiary care center equipped to handle complicated patients with rare diseases.
Pain
It is very hard when a child cannot express him or herself to know whether the crying is from pain or frustration. Children may have ear infections, toothache, aches and pains in their joints or feel discomfort from a full stomach. Do not hesitate to ask your doctor to check whether there is a physical reason for your child’s distress.
Palliative Care
Palliative care is any form of medical care or treatment that concentrates on reducing the severity of disease symptoms. The goal is to prevent and relieve suffering and to improve quality of life for people facing serious, complex illness. This support encompasses aspects such as respite care, symptom management and bereavement support and many extend over a period of time. An assessment of medical need and a care plan can lead to support provided to the child and family so both can experience a better quality of life.
Physical appearance
In ML II birth weight and length are often below normal. Clinical features of the disease are apparent at birth or in the first few months of life, including multiple joint contractures and kyphosis or scoliosis. In children with ML II, the neck is short, cheeks are often rosy, and the nose may be broad with a flattened bridge and upturned nostrils. The mouth may be wide, and children with ML II have very thickened gums. Eyebrows may be bushy and meet in the middle, and the eye sockets are usually shallow making the eyes appear prominent. Infants with ML II may have an unusual head shape, with prominence of the forehead. Some children with ML II may have fair skin, blonde hair and blue eyes, despite the colouring of their parents.
Physical Therapy
 Joint stiffness is a common feature of ML II. Limitation of motion and joint stiffness can cause significant loss of function. Range-of-motion exercises (passive stretching and bending of the limbs) may offer some benefits in preserving joint function, and should be started early. Passive hydrotherapy is beneficial for the joints while minimizing painful impact. Exercises that cause pain should be avoided. Once significant limitation has occurred, increased range-of-motion may not be achieved, although further limitation may be minimized.
Joint stiffness is a common feature of ML II. Limitation of motion and joint stiffness can cause significant loss of function. Range-of-motion exercises (passive stretching and bending of the limbs) may offer some benefits in preserving joint function, and should be started early. Passive hydrotherapy is beneficial for the joints while minimizing painful impact. Exercises that cause pain should be avoided. Once significant limitation has occurred, increased range-of-motion may not be achieved, although further limitation may be minimized.Skin
Taking a break
Caring for a severely affected child is hard work. Parents need a break to rest and enjoy activities, which may not be possible when their affected child is with them. Brothers and sisters also need their share of attention and need to be taken on outings that may not be feasible for their affected child. Many parents use some form of respite care or have someone come in regularly to help at busy times.
Additional Resources and Support Groups
Anesthetic Issues for Children with Mucolipidosis
The following information is provided as a public service by ISMRD and the Greenwood Genetic Center in Greenwood, S.C. This alert is co-authored by Sara Cathey, M.D. and Jules LeRoy, M.D., Ph.D. ISMRD is very grateful to the authors for the time they invested to make this information available to families with this diagnosis.
If you require further information about this medical alert you may contact ISMRD or Sara Cathy, M.D. at the Greenwood Genetic Center. This information is also available as a PDF file, but make sure you have the latest version of Adobe Acrobat first! If you do not have Acrobat, then download it from the Adobe website.
Medical Alert for Parents of Children with I Cell Disease and the Physicians Who Care for Them
I Cell disease is a slowly progressive inborn error of metabolism for which, unfortunately, no curative treatment is available. Children with I Cell disease have extreme growth failure, neurodevelopmental delay and disease of connective tissues in many organs, including the lungs and respiratory tract. Manifestations including swollen gums, stiff joints, thickfeeling skin, bone and tendon abnormalities are well known signs of the disorder.
Because of the poor growth potential, atrophied muscles, and poor compliance of the narrow thoracic cage, the baseline respiratory status of these children is compromised. Additionally, they have small mouths that do not open fully or easily. As the children grow older the airway becomes gradually more restricted as the initially swollen airway passages become stiff and easily traumatized. This combination of factors makes intubation (placing a tube within the airway in order to use mechanical assistance to breathe for the patient) very risky, so elective surgical procedures should be avoided as much as possible.
If a procedure is considered essential, it should be undertaken at a major medical facility where pediatric anesthesia and pediatric critical care services are available.Under all circumstances,medical staff should be prepared to perform fiberoptic intubation, where a tiny camera is used to visualize the airway during the intubation procedure. Because children with I Cell disease have much smaller airways than other children of the same age, a much smaller endotracheal tube will be required. However, a smaller tube may be less effective for ventilation (breathing for the patient) and may potentially lead to additional problems such as pneumothorax or collapsed lung.
All medical personnel involved with the elective procedure (surgeons, anesthesiologists, and critical care specialists) should be conversant with the clinical features and natural course of I Cell disease. Parents and physicians must realize that intubation can easily cause more problems than an elective procedure may alleviate.
For many of the same reasons already described, extubation (removing the breathing tube) may also be unusually difficult andthe patient’s general condition in the first days following it, quite unpredictable. If prolonged intubation is required, a tracheotomy (making an artificial hole in the trachea through an incision in the neck) will be required. The probability of subsequent adequate healing in order to close the tracheostomy and having the patient breathe without mechanical assistance is slim indeed.
All humans have an airway protective reflex so as to continue to breathe when sleeping or even when artificially sedated. Deep anesthesia diminishes this reflex. Children with I Cell disease do not necessarily respond differently to anesthesia. However, because of their precarious airway status from the start,the patients may lose that protective reflex very quickly, even from“light sedation”. That is why a medical team must always be prepared fo rintubation, preferably fiberoptically, if indeed a surgical procedure isc onsidered of crucial importance.
Sara Cathey MD
Jules G. Leroy MD. PhD
Greenwood Genetic Center, Greenwood, South Carolina, USA
Feeding and Nutrition in children with ML II alpha/beta
Living and caring for a Mucolipidosis II alpha/beta child comes with many challenges. A group of families who have Mucolipidosis II alpha/beta children have put together their experiences of caring for their children in the fact sheet below. Please be aware that these are family experiences only and long term care of your child must be under the guidance of your treating doctor.
Feeding and Nutrition in children with ML II alpha/beta suggestions for feeding a child with I-Cell Disease – A family experience. (View or download PDF)
Patient Support Groups:
- ISMRD is the international support group for Alpha-Mannosidosis
- ISMRD’s Facebook page; this is our closed group for families only
- ISMRD’s public Facebook page for this disorder
- • The National MPS: Society based in the United States
- • The Canadian MPS Society
- • The UK MPS Society
 image")
Medical and Research Information
- Mucolipidosis review by Dr. Richard Steet. Prepared in 2016 and updated 2018 – Review here
- GeneReviews: Gives good descriptions of genetic diseases. Information is comprehensive, and access is free to all users.
- Genetic Home Reference: An excellent description of Mucolipidosis II alpha/beta
- GeneTests: List of laboratories testing for Mucolipidosis II.
- Clinical Trials Registry of federally and privately supported clinical trials conducted in the United States and around the world. This site gives information about each trial’s purpose, who may participate and the location.
- OMIM: Technical information about the genetics of Mucolipidosis II. OMIM is a site developed for scientists and medical specialists and contains both general and highly technical information. Access to this site is free.
ML III alpha/beta (Pseudo-Hurler Polydystrophy)
Mucolipidosis III alpha/beta results from a deficiency of the enzyme N-acetylglucosamine-1-phosphotransferase. Just as luggage in an airport is tagged to direct it to the correct destination, enzymes are often “tagged”. In Mucolipidosis III alpha/beta, the deficient enzyme is supposed to tag other enzymes (activator proteins) so that they can initiate certain metabolic processes in the cell. Because the activator proteins are not properly tagged, they escape into spaces outside the cell and therefore cannot do their usual work of breaking down substances inside the cells.
Mucolipidosis III alpha/beta is a slowly progressive disorder that affects many parts of the body. Signs and symptoms of this condition typically appear around age 3 or 4, but some children are not diagnosed until later on in life.
Individuals with mucolipidosis III alpha/beta grow slowly and have short stature. They also have stiff joints and dysostosis multiplex, which refers to multiple skeletal abnormalities seen on x-ray. Many affected individuals develop low bone mineral density (osteoporosis), which weakens the bones and makes them prone to fracture. Osteoporosis and progressive joint problems also cause bone pain that becomes more severe over time in people with mucolipidosis III alpha/beta.
How Common is it?
Mucolipidosis III alpha/beta is a rare disorder, although its exact prevalence is unknown. It is estimated to occur in about 1 in 100,000 to 400,000 individuals worldwide.
The A to Z of Mucolipidosis III
Anaesthetics
Anaesthesia, although very safe, must be well planned. It is important to spend time with the anaesthetic team talking through some of the problems these children have that may complicate the use of anaesthesia. These issues may include:
- Short Necks
- Limited jaw and neck movement
- Thick tongues
- Airways obstructed by storage material
- Floppy tracheas (windpipes)
- Inefficient breathing. Abnormalities of the spine and ribs can limit the patient’s capacity to breath and fully expand their lungs.
Anaesthesia in young children should always be performed by a paediatric anaesthesiologist who has experience with difficult airways and skeletal dysplasias. The neck of these children may be somewhat lax and could be more prone to injury during intubation. Also, the storage material may interfere with the placement or removal of the tube. Very small endotracheal tubes may have to be utilized. However, under the correct supervision, surgeries most often go very well.
In older ML patients a safer approach to passing a tube into the airway (intubation) might be by numbing the mouth and throat with local anaesthetic and passing a fine flexible telescope into the wind-pipe (trachea) while the person is a wake and co-operative. Then an endotracheal tube can be passed over the telescope and the airway secured before the person is put to sleep.
The Australian MPS Society has a wonderful booklet called MPS and Anaesthesia it can be ordered by contacting them on their website.
Carpel Tunnel Syndrome / Hands
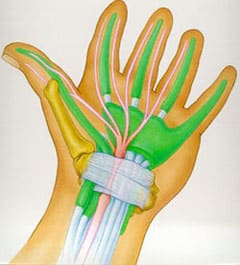 Carpel tunnel syndrome is very common in MLIII. The clinical signs are sensations of “pins and needles” in the fingers, loss of feeling and wasting of the muscle at the base of the thumb. The hand could also be weak.
Some children will never complain of pins and needles. They may have had symptoms for a long time, but do not understand what is meant when questioned regarding pain because of the constant nature of the pain sensations they have been experiencing. You may notice that the child will not be aware of hot and cold. This is an indication that perhaps there is nerve compression in the carpel tunnel.
To determine if the child has this compression, nerve conduction tests are necessary to check for nerve damage. This test feels like small spiders running up the hand and lower part of the arm. Most children tolerate this test really well but it has been noticed there can be moderate pain near the end of the testing process.
Carpel Tunnel has been diagnosed in an ML III child as young as 4 years of age.
Many doctors will doubt that young children have this condition. You will need to be persistent and explain that your child has a storage disorder and this is a frequent finding associated with the condition.
The median nerve travels from the forearm into your hand through a “tunnel” in your wrist.
The bottom and sides of this tunnel are formed by wrist bones and the top of the tunnel is covered by a strong band of connective tissue called a ligament.
This tunnel also contains nine tendons that connect muscles to bones and bend your fingers and thumb.
These tendons are covered with a lubricating membrane called synovium which may enlarge and swell under some circumstances.
If the swelling is sufficient it may cause the median nerve to be pressed up against this strong ligament which may result in numbness, tingling in your hand, clumsiness or pain.
Another important aspect to consider with Carpel Tunnel in these children is that although the carpel tunnel will be tight and will be compressing the nerves travelling through this area, compression can also be found either higher into the wrist area or down into the palm of the hand. It is important to explain this to your doctor and ask him to look further than the Carpel Tunnel. Remember, your child has a storage disease that affects the entire body.
Carpel tunnel syndrome is very common in MLIII. The clinical signs are sensations of “pins and needles” in the fingers, loss of feeling and wasting of the muscle at the base of the thumb. The hand could also be weak.
Some children will never complain of pins and needles. They may have had symptoms for a long time, but do not understand what is meant when questioned regarding pain because of the constant nature of the pain sensations they have been experiencing. You may notice that the child will not be aware of hot and cold. This is an indication that perhaps there is nerve compression in the carpel tunnel.
To determine if the child has this compression, nerve conduction tests are necessary to check for nerve damage. This test feels like small spiders running up the hand and lower part of the arm. Most children tolerate this test really well but it has been noticed there can be moderate pain near the end of the testing process.
Carpel Tunnel has been diagnosed in an ML III child as young as 4 years of age.
Many doctors will doubt that young children have this condition. You will need to be persistent and explain that your child has a storage disorder and this is a frequent finding associated with the condition.
The median nerve travels from the forearm into your hand through a “tunnel” in your wrist.
The bottom and sides of this tunnel are formed by wrist bones and the top of the tunnel is covered by a strong band of connective tissue called a ligament.
This tunnel also contains nine tendons that connect muscles to bones and bend your fingers and thumb.
These tendons are covered with a lubricating membrane called synovium which may enlarge and swell under some circumstances.
If the swelling is sufficient it may cause the median nerve to be pressed up against this strong ligament which may result in numbness, tingling in your hand, clumsiness or pain.
Another important aspect to consider with Carpel Tunnel in these children is that although the carpel tunnel will be tight and will be compressing the nerves travelling through this area, compression can also be found either higher into the wrist area or down into the palm of the hand. It is important to explain this to your doctor and ask him to look further than the Carpel Tunnel. Remember, your child has a storage disease that affects the entire body.Central Nervous System Involvement
There is no real documentation on CNS involvement in ML III, it is thought that these children do not have CNS involvement, but there are several children around the world who exhibit this problem.
If your child is complaining of pins and needles in their arms and legs it is important that an MRI is done and it is read by a doctor who has a very good understanding of ML. If an MRI is done of the brain the doctor will see white patches in the brain this is storage material and can be mistaken for other disorders. Once again make sure you tell the doctor you child has a storage disease.
There are no easy answers to this problem in ML III, and requires further research.
Dental treatment
If your child has a heart problem (refer to the Heart section later), you will need to ensure that before any dental treatment is carried out such as scaling, fillings or dental extraction, they may need to take prophylactic antibiotics. The mouth has certain bacteria that may get into the bloodstream and cause infection in the heart valves.
If teeth need to be removed under anaesthetic, this should be done in a hospital situation under the care of an experienced anaesthetist.
Some ML III children do require dental extraction of their baby teeth as they tend not to fall out. Others will have problems with new teeth erupting through the gums and this process can be assisted by the Dentist under hospital procedure.
Diet
Doctors
Once your child has been diagnosed with ML you will need to work with your Paediatrician or Genetic Specialist to build a team of Doctors who will help you manage the many complications of ML.
Some of these Doctors could be:
- Cardiologist
- Dentist
- Orthopaedic specialist
- Endocrinologist
- Dietician
- Eye Specialist
- Ear Nose and Throat Specialist
- Neurologist
- Spinal Surgeon
Eyes
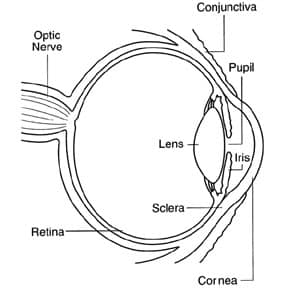 ML children need to have regular eye checks to look for clouding of the cornea.
The cornea is the eye’s outermost layer. It is the clear, dome-shaped surface that covers the front of the eye.
Because the cornea is as smooth and clear as glass but is strong and durable, it helps the eye in two ways:
1. It helps to shield the rest of the eye from germs, dust, and other harmful matter. The cornea shares this protective task with the eyelids, the eye socket, tears, and the sclera, or white part of the eye.
2. The cornea acts as the eye’s outermost lens. It functions like a window that controls and focuses the entry of light into the eye. The cornea contributes between 65-75 percent of the eye’s total focusing power.
In ML children, the corneas may become cloudy due to build up of storage material. If corneal clouding is severe, it may reduce sight. Additionally, light may come into the eye unevenly, especially bright lights. Thus sunglasses in bright lights may help. And, if vision is reduced, you should make sure there is appropriate lighting and avoid dim lights. For significant corneal clouding, causing limitation in vision, a corneal transplant may be considered.
ML children need to have regular eye checks to look for clouding of the cornea.
The cornea is the eye’s outermost layer. It is the clear, dome-shaped surface that covers the front of the eye.
Because the cornea is as smooth and clear as glass but is strong and durable, it helps the eye in two ways:
1. It helps to shield the rest of the eye from germs, dust, and other harmful matter. The cornea shares this protective task with the eyelids, the eye socket, tears, and the sclera, or white part of the eye.
2. The cornea acts as the eye’s outermost lens. It functions like a window that controls and focuses the entry of light into the eye. The cornea contributes between 65-75 percent of the eye’s total focusing power.
In ML children, the corneas may become cloudy due to build up of storage material. If corneal clouding is severe, it may reduce sight. Additionally, light may come into the eye unevenly, especially bright lights. Thus sunglasses in bright lights may help. And, if vision is reduced, you should make sure there is appropriate lighting and avoid dim lights. For significant corneal clouding, causing limitation in vision, a corneal transplant may be considered.Ears
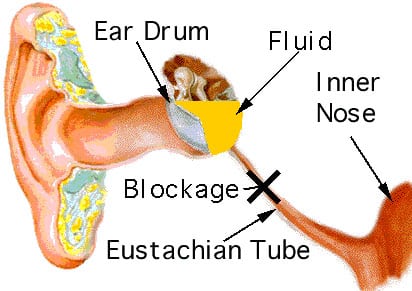 ML III children can suffer from many ear infections in their early childhood.
When the eustachian tube doesn’t operate correctly, due to anatomical problems, injury or swelling, new air is not able to reach the middle ear.
The body will remove the air from the middle ear and this creates a vacuum (negative pressure). When the pressure is mild, this may cause stuffiness or pressure in the ear.
When the pressure increases, the body senses pain. When the pressure remains for a period of time, the body replaces the vacuum with fluid. The fluid behind the eardrum can lead to problems with hearing loss and ear infections. If this negative pressure persists, fluid from the lining of the middle ear will build up and in time become thick and like glue. Hence the condition known as “glue ear”
Ventilation tubes (grommets, PE tubes) are frequently employed in the ML III population to manage chronic serous otitis media, or recurrent otitis media not responsive to medical management. Each child needs careful evaluation by an ENT specialist, and if surgery is recommended, patients should see the anesthesiologist (anesthetist in NZ, UK) beforehand to ensure that the risks associated with anesthesia have been addressed and minimized.
ML III children can suffer from many ear infections in their early childhood.
When the eustachian tube doesn’t operate correctly, due to anatomical problems, injury or swelling, new air is not able to reach the middle ear.
The body will remove the air from the middle ear and this creates a vacuum (negative pressure). When the pressure is mild, this may cause stuffiness or pressure in the ear.
When the pressure increases, the body senses pain. When the pressure remains for a period of time, the body replaces the vacuum with fluid. The fluid behind the eardrum can lead to problems with hearing loss and ear infections. If this negative pressure persists, fluid from the lining of the middle ear will build up and in time become thick and like glue. Hence the condition known as “glue ear”
Ventilation tubes (grommets, PE tubes) are frequently employed in the ML III population to manage chronic serous otitis media, or recurrent otitis media not responsive to medical management. Each child needs careful evaluation by an ENT specialist, and if surgery is recommended, patients should see the anesthesiologist (anesthetist in NZ, UK) beforehand to ensure that the risks associated with anesthesia have been addressed and minimized.Growth
Children with ML III may grow to between four foot six inches (140cm) and five feet (152cms), but once again there is a huge variation. Some ML III children are similar in size to MLII



Heart Involvement
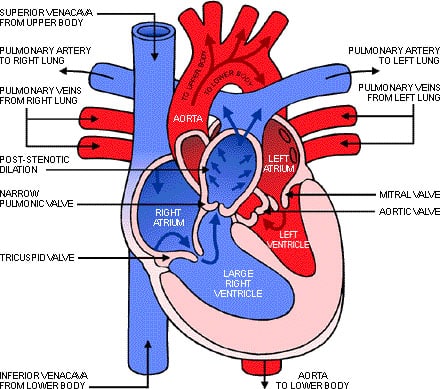 Heart murmurs will be heard by the doctor if your child has valves that have become damaged by storage material. Usually at this time your doctor will ask for an echocardiogram to be done. An echocardiogram is an ultrasound of the heart, much like the screening done when babies are in the womb. This allows the doctor to see what is happening to the heart
In ML III it is usually the Aortic and mitral valves that are causing problems, as the storage material builds up the valves become thickened and do not close off correctly.
The heart has four valves that open and close to keep blood flowing in the right direction through the heart.
The aortic valve connects the left ventricle to the aorta, which then allows blood to circulate to the rest of the body.
The mitral valve connects the heart’s upper-left chamber, the atrium, to the heart’s lower-left chamber, the ventricle.
When the valves do not function properly, amounts of blood may flow backward in your heart. This means that blood is not flowing as efficiently through the heart and to the rest of the body.
Storage of lysosomal material may weaken or thicken the valves, creating leakage around the valves. Treatment depends on the severity and progression of the valve’s leakage. It is possible that the leakage may not cause any problems; however it may cause fatigue and shortness of breath that may require surgery to repair or replace the valves.
Heart murmurs will be heard by the doctor if your child has valves that have become damaged by storage material. Usually at this time your doctor will ask for an echocardiogram to be done. An echocardiogram is an ultrasound of the heart, much like the screening done when babies are in the womb. This allows the doctor to see what is happening to the heart
In ML III it is usually the Aortic and mitral valves that are causing problems, as the storage material builds up the valves become thickened and do not close off correctly.
The heart has four valves that open and close to keep blood flowing in the right direction through the heart.
The aortic valve connects the left ventricle to the aorta, which then allows blood to circulate to the rest of the body.
The mitral valve connects the heart’s upper-left chamber, the atrium, to the heart’s lower-left chamber, the ventricle.
When the valves do not function properly, amounts of blood may flow backward in your heart. This means that blood is not flowing as efficiently through the heart and to the rest of the body.
Storage of lysosomal material may weaken or thicken the valves, creating leakage around the valves. Treatment depends on the severity and progression of the valve’s leakage. It is possible that the leakage may not cause any problems; however it may cause fatigue and shortness of breath that may require surgery to repair or replace the valves.Infections
During the winter months some ML III children get chest and respatory infections that require broad spectrum antibiotics. Many parents find the benefits of keeping their children on low dose antibiotics during these months to prevent infections setting in.
If your child has a cold or cough that does not seem to be getting better, you should seek medical advice.
Intelligence
Variation in IQ is considerable. Some children have normal intelligence, but some will have a learning disability which could involve short term memory loss. The reason for this is the continual build up of material being stored in the cell.
These children learn by repetitive teaching, and will require the services of special education, or teacher support time in mainstream education. To achieve the best education possible many of these children have IEP’s (Individual Education programs) which allows them to learn at their level.
An example of how the gap widens is seen in one ML3 young adult who is 23 years old. His learning level is 9 years of age and his reading level is 6 years of age, but he has an amazing memory for anything relating to sport. Early intervention was not part of this young mans life due to a late diagnosis of age 6. The informative years were lost and so the gap continued to grow over time.
It is important that a policy of early intervention be followed with the education of these children. Proactive education for these children will give access to necessary tools and materials and will increase a positive learning atmosphere and provide a guide base for each new teacher in your child’s future.
This requires that parents must become advocates for their children in fighting for services, therapies, and educational assistance. Laws and resources differ across the world, so it is important to learn about intervention services where you live.
Waiting and then establishing a reactive IEP will lose valuable time in your child’s overall education.
An example of what early intervention can do is seen in an 18 year old girl who was diagnosed at 2 years of age, by age 3
Life expectancy
Those with ML III can live well into adulthood, some being supported at home others living independently, or in supported living arrangements.
There are a small number of ML III children who are severely affected and do not have a long life expectancy. It is important to note the wide range of individual abilities as our young adults mature. Parents should be prepared to understand the diversity and range of their ML III child, so they can help prepare a better future for their child.
Living with ML III
Living with a child with ML III can certainly be a challenge, but also a joy. As parents of these very special children you will spend many hours learning everything there is to know about this disease, and over time you will become an expert in advocating for the best possible health care and Education available. It is important that parents actively have respite time away from their children to “re-charge” their batteries. You should not feel guilty about this but actively use this time as a tool to maintain your own energy levels and thus be a better advocate in the long term for your child.
One of the best gifts you can give your child is the gift of your own health as the caregiver and parent.
You may also wish to discuss the long term implications of care and funding for your child with your own legal adviser in the event that your own health or your pre-mature death will not allow you to continue caring and advocating for your child. There are many tools such as Trusts and Life Insurance that can be put in place to secure the long term future of your child.
Another useful tool is an Enduring Power of Attorney that can allow you to speak on behalf of your adult children for such things as health care, money management and rights to social security benefits. (In the United States it is referred to as Durable Power of Attorney and will differ from Country to Country)
Metabolic bone disease
Secondary Metabolic bone disease is very new to ML II and III. Very few doctors know about this and those that are introduced to it often state that this treatment will not work in ML.
The reason they say this is that once again bone disease in ML is not well understood and is often likened to Hurler disease.
The following information is written by the family who went through the first clinical trial with their two children and should be used along side the Medical paper, The Osteodystrophy of Mucolipidosis type III and the effects of intravenous Pamidronate Treatment, and Protocols for Use of Pamidronate, which can be downloaded as an Adobe PDF file.
Permission has been kindly given to us by Kluwer Academic Publishers who hold the copy rights.
(Kluwer Academic Publishers has merged with Springer-Verlag; the two publishing companies are now operating under the joint Springer brand. As a result, this Kluwer website has been redirected to springeronline.com)
Common Orthopaedic complications in ML III
Patients suffer to varying degrees the following complications.
- Stiffness of hands and shoulders
- Claw hand deformity
- Scoliosis
- Short stature
- Low iliac wings, shallow acetabular fossa
- Erosion of the femoral heads
- Valgus deformity of femoral neck
- Underdevelopment of posterior elements of dorsal spine
Dysostosis multiplex
- Skull: calvarial thickening, premature suture closure, shallow orbits, abnormal tooth spacing, J-shaped sella
- Spine: anterior hypoplasia of lumber vertebrae, kyphosis
- Long bones: shortened, wide diaphyses, irregular metaphyses, poorly developed epiphyseal centres
- Clavicles: short, thickened, irregular
- Ribs: narrow at vertebral ends, flat and broad at sternal ends
- Phalanges: shortened, trapezoidal in shape, widened diaphyses
What is happening to the bones of ML Children?
We all have cells that lay down new bone and cells that break down old bone; this is an on going process in all of us.
The cells that lay down new bone are called Osteoblasts and the cells that break down old bone are called Osteoclasts.
In children with Mucolipidosis type II & III the Osteoclast activity is extremely elevated and the osteoblast activity extremely low. So in effect ML children actually break down more bone than they lay down.
The nature of the bone pathology is that bones are extremely brittle and crumbling. Some degree of fracturing around the joints is inevitable from time to time. Each episode of fracturing and crumbling results in considerable pain and in some cases a loss of mobility.
An example of elevated osteoclast activity is seen in this example below.
The Normal range for bone turnover is <120 and is appropriate for age and sex.
Sibling One
| Time (months) | Alkaline Phosphatase |
| 0 | 256 |
| 6 | 187 |
| 12 | 156 |
You can see from here that sibling one at 18 years of age had extremely elevated bone turnover prior to treatment. These results are 6 monthly intervals where you can see a drop in bone turn over.
At four years of treatment you can see that the Osteoclast activity has dropped considerably.
| Date | Alkaline Phosphatase |
| 8/4/2004 | 93 |
| 5/5/2004 |
89 |
| 2/6/2004 |
90 |
Sibling two was considerably worse and was also going through puberty at the commencement of treatment.
Once again the Normal range is <250 and is appropriate for age and sex
Sibling Two
| Time (months) | Alkaline Phosphatase |
| 0 |
453 |
| 6 |
316 |
|
12 |
237 |
After four years of treatment you can see once again that Osteoclast activity has been reduced.
| Date | Alkaline Phosphatase |
| 8/4/2004 |
95 |
| 5/5/2004 |
89 |
| 2/6/2004 |
93 |
What is Pamidronate and how does it work?
Pamidronate is one of the many Bisphosphonates available to treat pain and bone disease. It is also known as (Pamisol /Aredia), and is well known in the treatment of Osteogenous imperfecta (Brittle Bone Disease)
When Pamidronate is used, the Osteoclast (bone reabsorption cells) activity is decreased allowing the Osteoblast (laying down of new bone) activity to take over, and so increase bone density and repair some of the bone damage.
In these two young people it was found though bone biopsies that the Perosteal surface (hard outer layer of bone) was not being changed by Pamidronate, but at the Endosteal and Trabacular surfaces (soft internal parts of the bone) there was significant change in bone density after 12 months of treatment.
Pamidronate in Mucolipidosis is given through IV at monthly intervals and has not been trialled orally. The reason for this is that ML children can have stomach problems with many different kinds of drugs.
Side effects of Pamidronate treatment
Most children will suffer some of the following symptoms in the first 2 infusions.
Flu like symptoms which include –
• Fever
• Nausea
• Vomiting
• Headaches and body aches and pains
These symptoms usually occur following the 1st infusion within 12 – 24 hours and subside within 48 hours. Some children who have commenced treatment in the United States reported body aches and pains which lasted for up to 7 days with the first infusion. These were managed with over the counter pain relief.
With subsequent infusions these symptoms do not usually occur but if they do it is to a lesser degree. Ibuprofen (Nurofen) and Paracetamol (Panadol) can be used to relieve any side effects.
Some of the children using this treatment have been broken in slowly to reduce the side effects of treatment. For example commencing with a ¼ dose in the first infusion ½ dose in the second infusion and a full dose in the 3rd infusion.
It has been noted that the side effects are less and the child recovers quickly from the 1st infusion.
Why was this chosen as a possible treatment for Mucolipidosis?
The two children from New Zealand who commenced the Study had the following problems prior to treatment.
• The older sibling at age 18 was suffering from significant hip and back pain, which was seriously compromising his quality of life, waking him from sleep and interfering with wheelchair transfers.
• The younger sibling by age 12 was also suffering from significant, constant joint pain, mainly from the hips. This was accompanied by a dramatic deterioration in her mobility. By age 14 she was unable to walk and was wheelchair bound.
• Many forms of analgesia had been used in an attempt to decrease their hip and back pain. These had all been unsuccessful.
•It was thought that these children could benefit from this treatment that showed outstanding results in Osteogenesis Imperfecta.
Treatment was commenced on 20th August 2000 with very rapid improvements
• 2 weeks after the first infusion both children reported they were pain free.
• The older sibling had increased mobility and was able to do all his own transfers.
• By 18 months of treatment he was able to stand and take four steps. He also has not had any chest infections since commencing treatment.
• The younger sibling also showed dramatic improvement in mobility. By 3 months of treatment she was able to walk with a walking frame and later progressed to Crutches and being able to walk unaided.
• Both siblings are now able to do many of their own day to day cares.
Bone Density Results during Treatment
Sibling One
| Scan Date | Age | BMD (g/cm2) | Z Score | Change % |
| 16/8/00 | 18.3 | 0.523 | -3.9 | ….. |
| 13/2/01 | 19.8 | 0.554 | -2.9 | 5.9% |
| 28/8/01 | 20.8 | 0.605 | -2.3 | 15.7% |
| 26/2/02 | 21.3 | 0.638 | -1.6 | 22.0% |
| 19/3/04 | 22 | 0.694 | -1.4 |
Sibling Two
| Scan Date | Age | BMD (g/cm2) | Z Score | Change % |
| 16/8/00 | 14.6 | 0.751 | -3.1 | ….. |
| 13/2/01 | 15.1 | 0.871 | -2.7 | 16.0% |
| 28/8/01 | 16.6 | 0.962 | -1.7 | 28.1% |
| 26/2/02 | 17.1 | 1.065 | -1.3 | 41.8% |
| 19/3/04 | 18 | 1.209 | 0.1 |
You can see that after four years of treatment bone density continues to increase.
What are the first steps to take leading up to treatment?
To create baselines from which progress of treatment can be monitored. The following tests are carried out prior to treatment
- Dexa Scan – this will measure bone density
- Full skeletal X-Rays with bone age taken on left wrist
- Renal Ultrasound
- Blood and Urine samples taken for Endocrine and Biochemistry value.
A full explanation for all these tests are explained in the Protocols which can be downloaded from ISMRD’s website
Metabolic screening
A screen for lysosomal diseases is undergone through a urine spot screen, which looks for the presence of accumulated storage material in the urine. However, some of the lysosomal storage diseases do not have positive spot screens. Or, more advanced spot screens have to be pursued. For example, many labs screen for Mucopolysaccharide build-up, but others screen for oligosaccharides. It is important to work with a medical professional that is knowledgeable in lysosomal diseases and screening to get an appropriate work-up. If screening comes back positive, doctors typically consider further testing based on the clinical presentation of your child. For many lysosomal disorders, this is done through a blood test or skin biopsy looking for a decrease in a specific enzyme. Typically, specific enzymes have to be ordered, thus a “panel” of lysosomal enzyme testing is not available.
A diagnosis of ML is made by showing that lysosomal enzymes are increased in the plasma of the blood. Because of the targeting defect in ML disorders, the enzymes are not in the lysosome, but are in the extracellular fluid. A ML disorder can be diagnosed by documenting an increase of lysosomal enzymes in the plasma of the blood. Further confirmation can be performed by looking at the enzyme activity of N-acetylglucosamine-1-phototransferase, which is decreased in individuals with ML. Other confirmation can be performed by looking for the specific gene changes (mutations) within this gene. Enzyme activity and mutation testing are not necessary to make the diagnosis of an ML disorder. However, if a family would consider prenatal diagnosis for a future pregnancy, this testing would need to be performed.
Mobility
With the large variation in ML III it is difficult to determine to what degree and when mobility becomes a critical issue. Children can suffer with mobility problems from as early as 4 – 5 years of age. There may be a noticeable change of gait to their walk. Long distances will become more difficult to achieve and the ML child may need to stop often for breaks in walking.
Other children could onset with independent walking at the ages of 9 – 10 years. With the onset of puberty there is a noticeable change in mobility. A mark able slow down in the ability to move independently for long periods increases to 5 or 10 minute walks, difficulty standing for long periods of time.
Metabolic bone disease must be considered at this stage as a major factor in the decline of mobility in your child. Wheelchairs and scooters, walking frames, crutches should be discussed with your doctor who will refer you to the appropriate people for assessment.
It is also important for parents to understand how these issues will cause difficulty in the schools and be prepared to have an IEP that addresses the appropriate measurements that need to be completed in order for your child to have success in school.
With this wide range – the onset of mobility issues and difficulty in walking may not occur till later into your young adult’s lives. This is determined by how each individual is affected by the build up of storage from this disease.
Pain
Many children with ML III will have chronic pain, which will interfere with their mobility, sleeping habits and quality of life.
Joint pain in Mucolipidosis will not respond to conventional drugs as we are not dealing with soft tissue damage in this disorder. We are dealing with secondary metabolic bone disease. Please refer to Metabolic Bone Disease for solutions to management of the pain issues.
Physical appearance
ML III is often not diagnosed in a child until around 4 – 6 years of age. During this time there are little to no signs of the disability.
One of the first signs that there is something wrong with your child may be the curling of the little fingers or stiff shoulder joints. He/she may start to walk or run differently from other children. Some children will also start to show signs of curvature of the spine (scoliosis); they may also have a barrel chest. All these symptoms need medical assessment.
Some children will also start to show signs of curvature of the spine; they may also have a barrel chest, short neck, roundness in lower cheek area. Legs my start showing signs of contractures. All these symptoms need medical assessment.
This is done by providing blood and Urine samples for analysis. Usually this is followed by a Skin Biopsy that is grown to fibroblast level where the level of enzyme activity can be looked at. This will give a true indication of which Lysosomal disease your child has.
By around age 9 it will start to become more noticeable that your child will be shorter than their peers, they may be unable to keep up with other children in the School playground. .
As the children get older there will be noticeable clawing of the hands. Some children will start having difficulty in putting their heals down when walking, this is due to contractures in their hips and knees becoming more evident, some will also start complaining of pain when walking long distances and begin holding onto their upper thigh, and needing to take rests more often. This is the time to start looking at wheelchairs and other mobility devices. The need to start protecting joints becomes more urgent at this age.
Physiotherapy/Physical Therapy as a treatment
Over the years there have been many discussions in regards to physio-therapy / PT and splinting with no clear reasons why one should or should not do this.
While the children are young and the disease process has not really kicked in they run, jump and play just like any normal child, this in effect could be called physo. As they child gets older their joints start to become stiff and they begin to slow down, mobility becomes a problem.
Many therapists believe that they are able to straighten out legs and arms and increase movement by giving these children physo. What has not been well understood in ML is that these children are beginning to have significant bone loss and physo of any kind should be incredibly passive. Physio-therapy / PT should not cause the child any pain. One must take great care of all ML joints, to prevent significant joint damage.
The best kind of physio-therapy for these children is Hydro-therapy. The warm water relaxes all the muscles and allows the child to have very relaxing passive therapy. Also, being in the water takes all the weight off their joints. Excellent results can be achieved by this kind of therapy.
Puberty
With the onset of puberty comes a major change in the chemical reactions within the body. This is normal for everyone. What this does though in ML is accelerate the rate at which some storage happens and also accelerates bone deterioration. This area has not been well understood or even recognised as something that plays a major roll in the deterioration and loss of mobility in ML III.
Through the bone studies carried out in New Zealand and Australia it has been reported by the families involved that around 9 – 10 years of age seems to be a critical time for ML III. Families reported that they were seeing their children deteriorate in mobility, quality of life, and an increase in pain.
Since the introduction of Pamidronate families in the United States are also pin pointing a marked change in their children from 9years of age.
Spine
Treatments
At present there is no cure for ML III. Many of the complications can be well managed by surgical procedures.
For ML II and ML III Pamidronate is now being used to treat Secondary Metabolic Bone Disease (please see Metabolic Bone Disease).
Additional Resources and Support Groups
Management. Treatment of manifestations: Low-impact physical therapy is usually well tolerated. Myringotomy tube placement should be used with caution in the treatment of recurrent ear infections. Carpal tunnel signs may require tendon release. In late childhood or early adolescence relief of hip pain following exercise becomes important; in older adolescents and adults with milder phenotypic variants, bilateral hip replacement has been successful. Later in the disease course management focuses on relief of general bone pain associated with osteoporosis, which has responded in a few individuals to monthly IV administration of the bisphosphonate pamidronate.
Prevention of secondary complications: Because of concerns about airway management, surgical intervention should be undertaken only in tertiary care settings with pediatric anesthesiologists.
Treatments: At present there is no therapy for Mucolipidosis III alpha/beta. Many of the complications can be well managed by surgical procedures. Secondary Metabolic bone disease can be well managed with Pamidronate. See medical papers below.
Patient Support Groups:
- ISMRD is the international support group for Alpha-Mannosidosis
- ISMRD’s Facebook page; this is our closed group for families only
- ISMRD’s public Facebook Page for this disorder
Various MPS societies around the world also support Mucolipidosis III
Medical and Research Information
- Mucolipidosis review by Dr. Richard Steet. Prepared in 2016 and updated 2018 – Review here
- GeneReviews: Gives good descriptions of genetic diseases. Information is comprehensive, and access is free to all users.
- Genetics Home Reference: This has an excellent description of Mucolipidosis III alpha/beta with good reference to other information and resources.
- GeneTests: A list of laboratories testing for Mucolipidosis.
- Clinical Trials: A registry of federally and privately supported clinical trials conducted in the United States and around the world. This site gives you information about the trial’s purpose, who may participate and the locations
- Orphanet: A database dedicated to information on rare diseases and orphan drugs. Access is free of charge.
- OMIM: Has technical information about the genetics of this disorder . It is a site developed for scientists and medical specialists and contains both general and highly technical information. Access to this site is free.
- Genetic and Rare Diseases Information Center (GARD): A collaborative effort of two agencies of the National Institutes of Health, The Office of Rare Diseases Research (ORDR) and the National Human Genome Research Institute (NHGRI) to help people find useful information about genetic conditions and rare diseases
Medical papers




Meet People with Mucolipidosis alpha/beta

Madison
Madison’s mum April has put together a video in tribute to her beautiful daughter and is thrilled to be able to share it with other families. Watch the video here.
Sam, Jesse-Rose and Damian
Meet three young ML III patients (Sam, Jesse-Rose and Damian) and their thoughts on how they live, what health problems they have, fears, desires… It is very unlikely that their story and thinking will not touch you.

Sam
I’m Sam and I was diagnosed with ML when I was about 6/7.
As I went through my teenage years my health steadily began to deteriorate. I attended numerous hospital appointments for X rays, scans, physio, psychological assessments for school etc. ML affects most joints and organs of my body.
By the time I was 15, I’d had carpal tunnel, 8 Plates, dental surgery and two full hip and socket replacements with more surgery to follow as the condition deteriorates with age.
I’m 19 now and live a fairly independent life living with my family. I’m a full-time wheelchair user and I travel independently on public transport. I volunteer at the National Football Museum, Lowry theatre and with a Special Needs Sports group.
Although I’m quite happy and content, I wouldn’t wish this disease on anyone. I have missed out on so much that my friends are able to do, but I’m not bitter. We need to promote awareness of rare diseases and try to find a cure and therapies to help us.
His mother Shirley describes one event from everyday life:
One day we went out for some fresh air to a park we’ve been going to for years. There was a very tall slide in the playground, probably three sections/platforms high. Sam said he had climbed to the top many times when he was younger, but now he wouldn’t get past the first rung. I asked if he felt sad or bitter about the memory? He said ‘No’ it’s just the way things turned out.

Matas
I’m Matas and I was born in 2010 in Lithuania. At 2 ½ years of age, I was diagnosed with ML ll/lll.
After this I had regular hospital appointments. This diagnosis was a shock to the family, as they had never thought that there was something wrong till then. It was especially difficult because there was so little information and no cure.
I’m very cheerful, friendly, curious and careful. I have a little brother and a dog and I love them both very much.
Despite moving difficulties, I like to play table tennis and football. ML ll/lll mainly affects my bones, muscles, lungs and other organs. I might be shorter in height than others but they say I have a very big heart and that I’m very strong as well.
I like to say “Don’t worry, such is life”.
My family loves me to the moon and back and I’m very happy to have them. ❤

Hayden and Sarah
Jenny Noble, from Tauranga New Zealand recounts their family’s experience with Mucolipidosis III alpha/beta, and details the dramatic improvements to Hayden and Sarah’s health and quality of life, following a trial of Pamidronate to treat the secondary bone disease that became such a significant problem in their teenage years.

Damien and Jesse-Rose
Juanita talks about Damian and Jesse-Rose’s journey to diagnosis and beyond. She also says, If anyone asks me emotionally how we have been going, I let them know we are positive and doing what we can to make the kids lives easier. It’s hard to say out loud that there are times when I feel devastated, times when I feel like I could fall into a dark pit and never come out again. There are many times when I feel powerless in changing anything, so I pray! I pray that we find at the very least a treatment that means they have minimal pain, that they can have functioning useful lives and contribute to society. I pray mostly that we find a cure.
Whatever our journey is, it will be one that is perhaps more heartfelt and precious than it might have been prior to receiving this diagnosis.