real life accounts
Family Stories
Hear from members and families of ISMRD as they share their experiences. Some of these were written specially to celebrate Rare Disease Day in 2022, and to spread awareness of Glycoprotein Storage Diseases.
Share your personal or family story by submitting our form below.
Curious about a specific disease? Click below to jump directly to family and personal stories for each listed disease.
Alpha-Mannosidosis | Aspartylglucosaminuria | Beta-Mannosidosis | Fucosidosis | Galactosialidosis | Mucolipidosis II alpha/beta | Mucolipidosis III alpha/beta | Mucolipidosis III Gamma |Sialidosis
Alpha-Mannosidosis

Sarah
Sarah and her family’s journey with Alpha-Mannosisosis is presented in an educative and positive light.
Sarah has written about her experience with Alpha Mannosidosis in her blog and includes updates about how her life is post-bmt. To read her blog, visit AchieveTheImpossibleToday.com.

Taryn
This is Taryn’s story about Living with Alpha-Mannosidosis and how the family of a teenager with a rare genetic disease copes with the affects of the disorder and how she gets her wish to meet her idols.

Timothy & Hollie
Our son and daughter, Timothy and Hollie are twins, born 14 November 1974. They were premature (about one month). There were some early indicators that it was more than just a rough start for them, but nothing really clear till they were about one year old and a number of milestones were not met on time.

Robert
This is the ongoing journal of Robert Stark’s fight against Alpha-Mannosidosis via Bone Marrow Transplant. Families and caregivers of children with mannosidosis and other Lysosomal Diseases face complex decisions about the future. It is the hope of Robert’s parents, Kathleen and Mark, that the pages within will prove helpful when such decisions must be made.
This is Robert’s Road to Recovery after diagnosis and Bone Marrow Transplant

Luke
Luke was diagnosed with alpha-mannosidosis when he was 5. He underwent a bone marrow transplant in September 2010, 5 months after being diagnosed. We believe the bone marrow transplant has helped treat some of the symptoms of this disorder.
This is Luke’s Blog written by his family as he underwent Bone Marrow Transplant.

Saffron
Saffron's Blog written by her parents documenting her journey through Bone Marrow Transplant.


Presley
I was diagnosed at age 3, with alpha-mannosidosis, had a bone marrow transplant at age 4. Some hobbies are hanging out with family and friends, arts and crafts, and playing with my dogs.
Being born with a rare disease means to me that even though I was born different, I am just like everyone else.
I am a host at Rafferty’s in Bowling Green, Kentucky and I am the founder of Presley’s Promise Homeless Outreach.
I have difficulties with being small, standing for long periods of time, some processing and some social awareness & some social skills.

Ville
Ville was diagnosed with Alpha-Mannosidosis in 1996 at the age of six years. The diagnosis helped Ville to have physical, speech and psycho therapies and he started his school at a special class. For a few years, we tried to find more information about the syndrome and any treatment.
How happy I became when I was contacted by Mr. Paul Murhpy who worked hard to found the ISMRD! At that time, there were only 5-6 affected persons in Finland, so we had no peer affected families in our country. Through the community of the ISMRD where I also was a board member for a couple of years, we networked with other families with affected children all over the world, at first by email, then through the ISMRD webpage and later, the Facebook group. It has been most important to be a member of this ISMRD community to share experiences, feelings and stories. In May 2002, we were happy to join the world-wide activities of the ISMRD community by organising a flea market in our home town Oulu; the profits were donated to the ISMRD. I felt so good to support the society and to know that my work had some meaning for promoting research and connecting people.
In June 2002, our family participated in the international symposium on MPS and related diseases, taking place for four days in Paris. We had an excellent opportunity to make acquaintance with many interesting people, families and learned more about future perspectives of treatments from professionals. We met many active parents of affected children like Judith and John Forman and Dr. Dag Malm, some to mention. Later, Dr. Malm contacted me, inviting Ville to take part in his large research. We visited Tromso University Hospital three times yearly in 2007-2009. In the thorough studies, Ville was found to be affected by a very mild type of Alpha-Mannosidosis. A very good news!
Nowadays Ville is 35 years old and doing just fine. He works at a workshop for disabled people for three days per week and goes to a club house in other weekdays. Unfortunately, although the ERT has been lauched for commercial use in Europe, the Finnish authorities find the treatment too expensive for the benefits at his age. However, Ville enjoys every day: a lot of traveling and walking and cycling!

Layla
Layla was diagnosed with Alpha-Mannosidosis at age three after we discovered her hearing loss. When she was developing, she was meeting her milestones, but not exactly like other kiddos. While we had seen loads of doctors and therapists and even a geneticist, no one put the pieces together. It was a speech therapist who suggested to me there could be neurological issues that really set off my intense digging. Once we had a diagnosis, for us, the right choice was bone marrow transplant. The amazing team at University of Minnesota Children’s Massonic helped us through the entire process. Their kindness and professionalism is bar none.
A year and a half post BMT, Layla is doing amazing! She has amazing balance, is starting to ride a bike, her hearing has come back, and she is talking up a storm. She is still going to a special school for the deaf and hard of hearing, had multiple therapies a week to include enzyme replacement therapy, and has a ways to go still, but is also doing a lot of normal kid stuff like ballet, swimming and singing in the choir. She loves dress up and ice cream. We are so proud of her!

Kate
Kate was diagnosed with alpha-mannosidosis April 25, 2023. I think we probably all remember exactly where we were and what we were doing when we got the call that day from her geneticist who said, “this is what she has, I don’t know much about it, google it and then we can talk tomorrow if you have any questions.” It didn’t feel real. While this doctor was very compassionate, she simply didn’t know and the unknown felt incredibly overwhelming and scary to us. Prior to her diagnosis, Kate had issues with immune deficiency and hearing loss. She was wearing hearing aids and making good progress with IViG treatment. We thought the hearing loss was related to frequent ear infections which we thought could be explained by the immune deficiency. It was through the grace of God and her most astute pediatrician who made the recommendation for genetic testing because she just could never pin point where the hearing loss came from. Thankfully, her pediatrician left no stone uncovered.
As soon as she was diagnosed, Kate’s aunt delved into a research frenzy and became a fierce advocate for Kate and her parents. One of the articles uncovered in this research was by Dr. Steven Walkley highlighting his work and discovery of how a bone marrow transplant can stop the progression of alpha-mannosidosis. Taking a chance, but wanting to do everything in her power to fight for Kate, Kate’s aunt reached out to Dr. Walkley by googling his email address. Miraculously, he responded right away! A phone call was arranged and in speaking with Dr. Walkley, he was so compassionate and knowledgeable, providing a wealth of information about alpha-mannosidosis and the process of how a bone marrow transplant could help. He also gave very valuable insight based on his years of research and experience in cases of alpha-mannosidosis where someone received a BMT vs someone who had not. His insight was so valuable, and his expertise and knowledge so comforting. Up until this point, at every other turn, even at well-known pediatric medical centers, we were being told “just wait and see, she looks fine.” One of the metabolic specialists we had met with even asked Kate’s aunt if she was a physician because of how much she knew about alpha-mannosidosis and the questions she was asking, and this was just after a few weeks of research! Ultimately, this place did not have sufficient answers for our questions or a treatment plan for Kate that we felt comfortable with. Dr. Walkley had recommended Dr. Troy Lund at University of Minnesota, and by this time, we had also connected with the Melone family, whose daughter Layla was currently a patient at UM for a BMT for alpha-mannosidosis. We had also been able to speak with the Forsman family, whose daughter Sarah is a thriving young adult after her bone marrow transplant some 20 years ago. Our first interaction with University of Minnesota was on a phone call with Dr. Paul Orchard. In another series of many miracles for our Kate, this call had been arranged by a good family friend’s son who years ago had worked under Drs. Lund and Orchard for a fellowship. Like Dr. Walkley, we were immediately put at ease by Dr. Orchard’s expertise, insight, and thoughtful guidance and care. Dr. Orchard connected us with the bone marrow transplant coordinator at University of Minnesota and from there, things really started rolling along quickly. We traveled to Minnesota for a workup week and while nerve racking and overwhelming, at the same time, it was such a relief to be in a place where people have heard of the rare disease your child has and can provide expert care. It was during this week that we met with Dr. Troy Lund who would be leading Kate’s treatment team. Again, his expertise, laid back demeanor, and confidence helped to put our minds at ease that this was the best place for Kate to be. We were notified shortly after that they had found a 10/10 match for Kate through the Be the Match registry. The donor’s donation day was coordinated with Kate’s treatment and on 8/29/2023, Kate received her bone marrow transplant, Day 0. The time in Minnesota was not always easy. We were away from family and our 2 younger children, but we were blessed to be able to stay at Ronald McDonald House where we formed a home away from home and met friends who became like family. Families, like us, in similar situations with their children. There were good days and bad days, though mostly good days for Kate, for which we are so grateful. She responded beautifully to her treatment and did so well every step of the way. Day +100 came around Kate’s 6th birthday in early December and we were on our way home, back to Louisiana 12/14/2023. Since returning home, Kate is thriving. She has her yearly follow ups for monitoring and we are eternally grateful for the team at Minnesota for their ongoing care. Kate is now 7 years old, and she is happy and healthy! We have also been so blessed to have a full circle moment in Kate’s story; recently, the family was able to meet Kate’s bone marrow donor in person which was so special and also Dr. Steven Walkley and his wife, Rebecca, were able to travel down to Louisiana to meet Kate and family, another very special visit. A beautiful story of compassionate and expert care, patient advocacy, and the selfless gift of a stranger. Our lives are now forever woven together with Kate’s team. Kate affectionately calls them “her doctor, her scientist, and her donor.” To Kate’s doctors and nurses, her scientist, and her donor, thank you will never be enough! Kate’s crusade against alpha-mannosidosis has been a crusade that our whole community has rallied behind, we are so grateful. We continue to fight and advocate for Kate and all others with rare disease each and every day.
Aspartylglucosaminuria

Daniel and Alexander
It took us 16 years to learn our children’s correct diagnosis. Observing mine and other kids throughout many years, I came to believe that my sons’ delays were not all I’ve been told; there was something else standing between my kids and their ability to become what they want to be.

Karin
My name is Karin and I was diagnosed with Aspartylglucosaminuria in November of 1985.
I enjoy doing LiteBrite, coloring, 24 piece puzzles, watching movies, and many other activities.
My very favorite activities are taking long naps and petting my puppy.
Beta-Mannosidosis

Kendreona
My name is Kendreona and I’m a 12yr old from the USA. I love being outdoors, listening/dancing to country music, and shopping. Even though Beta Mannosidosis affects my life with daily physical and mental challenges I still loves to laugh and be the center of attention. LOL.

Skylar
A few months after Skylar was born, we noticed how peacefully she was sleeping during a Super Bowl game on tv. That’s when we knew she had hearing loss. After a visit to the geneticist and a few more tests, Skylar was diagnosed with Beta-mannosidosis a day before her 1st birthday. These past few years, she has struggled with developmental delays, Dysphasia, countless infections, severe burning in her hands and feet from peripheral neuropathy, vision and hearing loss and leukoencephalopathy (white matter changes to the brain).
Despite these challenges, she remains a beacon of light in our home. She loves to play with her older brothers, swing, climb, and roughhouse. She attends the Phoenix Day School for the Deaf and is quickly picking up on ASL. We are thrilled about The Lost Enzyme Project and a potential treatment in 2025
Fucosidosis

Nur
My name is Nur – I’m also known as Nunu or Noodlebug. I’m funny, cheeky and sassy and a total chatterbox.
I have an ultra rare title: I have two major diagnoses, one of severe Juvenile Idiopathic Arthritis and the other of Fucosidosis.
I had a double hip replacement when I was fourteen and I’m in a wheelchair, but I can still walk short distances although my Fucosidosis messes with my balance. I started college this year and I’m loving it.
In the past few years I had endured major operations, countless injections on a weekly basis, chemo, and these days a visit every month onto the wards at UCLH for infusions. I treat these appointments as social gatherings and hold court on the infusion suite.
In two weeks time I will once again prove that I’m a miracle, as I will surpass my predicted life expectation – in good health – as I will turn twenty years old.
Stephen & Lauren: We are siblings both with the ultra rare condition of Fucosidosis, but aren’t affected exactly the same.

Stephen
I’m 29 and struggle with joint stiffness, so I walk and move slowly in a very laboured way.
I don’t have a lot of stamina and get frequent colds and stomach upsets. The colds lay me low for quite a while, I can however do very limited occasional work at our local mail centre.
I enjoy watching sports but don’t play any.

Lauren
I’m 24 and I have much more stamina, recovering from minor illness very quickly.
I’m mentally much younger so I’m not allowed out on my own, I would be too vulnerable.
I have quite a lot of red marks, angiokeratomas on my body but they do not bother me.
I enjoy music and dancing
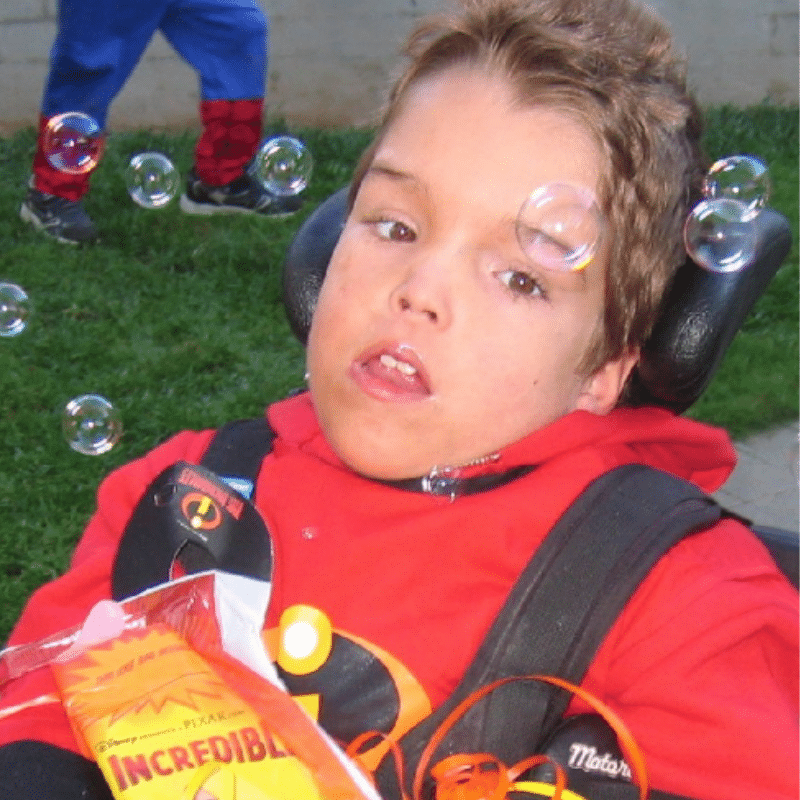
Matthew
Matthew was born a bright and happy boy, but his world was gradually clouded by a rare, degenerative disorder called Fucosidosis. This condition, caused by the absence of a vital enzyme, led to the accumulation of certain sugars in his body, slowly robbing him of his health.
As the disease progressed, Matthew’s once vibrant spirit was dimmed. He lost the ability to move and speak, and was plagued by frequent seizures. His frail body was further weakened by respiratory infections, eventually succumbing to a devastating bout of double pneumonia at the age of 10.
In 1998, when Matthew was first diagnosed, the internet was still in its infancy. Armed with only a few research articles, we felt utterly isolated. The weight of uncertainty and the fear of the unknown were overwhelming. Joining the International Society for Mannosidosis and Related Diseases (ISMRD) was a turning point. We found solace in connecting with other families facing similar challenges. The 2007 ISMRD conference in Michigan was a pivotal moment, where we met other Fucosidosis families and researchers working on potential treatments.
Galactosialidosis

Ivan the Great
My little “Ivan The Great” was diagnosed January 2011 with Galactosialidosis and am told it is EXTREMELY rare. I will never forget the day I found out his diagnosis and on top of that, that there is no cure or treatment for it. I felt like my heart was ripped right out from my chest. The whole world and how I perceived it changed instantly. I viewed the world and people in a whole new way.
Mucolipidosis II alpha/beta

Madison
Madison’s mum April has put together a video in tribute to her beautiful daughter and is thrilled to be able to share it with other families. Watch the video here.
Mucolipidosis III alpha/beta
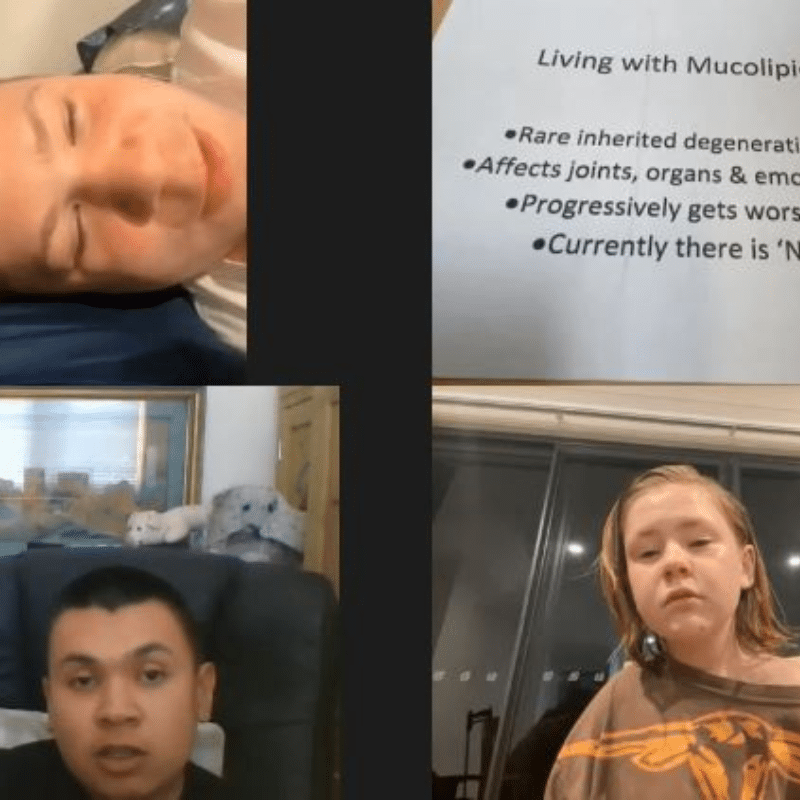
Sam, Jesse-Rose and Damian
Meet three young ML III patients (Sam, Jesse-Rose and Damian) and their thoughts on how they live, what health problems they have, fears, desires… It is very unlikely that their story and thinking will not touch you.

Hayden and Sarah
Jenny Noble, from Tauranga New Zealand recounts their family’s experience with Mucolipidosis III alpha/beta, and details the dramatic improvements to Hayden and Sarah’s health and quality of life, following a trial of Pamidronate to treat the secondary bone disease that became such a significant problem in their teenage years.

Damien and Jesse-Rose
Juanita talks about Damian and Jesse-Rose’s journey to diagnosis and beyond. She also says, If anyone asks me emotionally how we have been going, I let them know we are positive and doing what we can to make the kids lives easier. It’s hard to say out loud that there are times when I feel devastated, times when I feel like I could fall into a dark pit and never come out again. There are many times when I feel powerless in changing anything, so I pray! I pray that we find at the very least a treatment that means they have minimal pain, that they can have functioning useful lives and contribute to society. I pray mostly that we find a cure.
Whatever our journey is, it will be one that is perhaps more heartfelt and precious than it might have been prior to receiving this diagnosis.

Sam
I’m Sam and I was diagnosed with ML when I was about 6/7.
As I went through my teenage years my health steadily began to deteriorate. I attended numerous hospital appointments for X rays, scans, physio, psychological assessments for school etc. ML affects most joints and organs of my body.
By the time I was 15, I’d had carpal tunnel, 8 Plates, dental surgery and two full hip and socket replacements with more surgery to follow as the condition deteriorates with age.
I’m 19 now and live a fairly independent life living with my family. I’m a full-time wheelchair user and I travel independently on public transport. I volunteer at the National Football Museum, Lowry theatre and with a Special Needs Sports group.
Although I’m quite happy and content, I wouldn’t wish this disease on anyone. I have missed out on so much that my friends are able to do, but I’m not bitter. We need to promote awareness of rare diseases and try to find a cure and therapies to help us.
His mother Shirley describes one event from everyday life:
One day we went out for some fresh air to a park we’ve been going to for years. There was a very tall slide in the playground, probably three sections/platforms high. Sam said he had climbed to the top many times when he was younger, but now he wouldn’t get past the first rung. I asked if he felt sad or bitter about the memory? He said ‘No’ it’s just the way things turned out.

Matas
I’m Matas and I was born in 2010 in Lithuania. At 2 ½ years of age, I was diagnosed with ML ll/lll.
After this I had regular hospital appointments. This diagnosis was a shock to the family, as they had never thought that there was something wrong till then. It was especially difficult because there was so little information and no cure.
I’m very cheerful, friendly, curious and careful. I have a little brother and a dog and I love them both very much.
Despite moving difficulties, I like to play table tennis and football. ML ll/lll mainly affects my bones, muscles, lungs and other organs. I might be shorter in height than others but they say I have a very big heart and that I’m very strong as well.
I like to say “Don’t worry, such is life”.
My family loves me to the moon and back and I’m very happy to have them. ❤
Mucolipidosis III Gamma

Kelley
For many years after our daughter Kelley received the diagnosis of ML III in 1973, we weren’t able to connect with others sharing her diagnosis. By 2009, we joined the MPS Society and started to find others with Mucolipidosis, sharing phone calls and email messages.
I was excited when, in July 2003 I received a message from Paul Murphy, the subject line of which read, “ISMRD Welcomes ML III Families.”
That was the first time anyone welcomed us using the name of Kelley’s disease! Paul explained that, as the president of ISMRD, the conditions of ML II and ML III were a natural fit for the organization. In 2005, we were delighted to be able to meet other families at a Conference sponsored by ISMRD. Members traveled from all around the United States and as far away as New Zealand and Italy to attend “Crossing Oceans for a Cure.”
The Conference proved to be a wonderful opportunity for us to meet with others, share our stories, and gain knowledge from the doctors who presented valuable information that we would have had to travel far and wide to obtain.
Although Kelley died in 2009, at the age of 45, we continued to attend more conferences and met up personally with other families. The availability of Facebook has helped us stay in touch with our many friends in the community and we always look forward to updates from ISMRD.
Denise Crompton, author of DIAGNOSIS: RARE DISEASE
Sialidosis

Alessia
My name is Alessia and I am 11 years old. I live in the UK London; I like dance and art.
At the age of 3 I was diagnosed with Sialidosis after many tests and analyzes. At the age of 8, I was also diagnosed with epilepsy, starting treatment with Kepra.
My health started to deteriorate a year ago after I had Covid-19. I start using a wheelchair for a year and I am totally dependent on my mother or another adult.
Every day I try to enjoy as much as I can every day of my life and I fight with my whole life. I pray every day that God will heal me and give me strength. I hope that soon, we Sialidosis patients will be able to have a treatment that will make our lives easier.

Alexander
A Family’s fight against Sialidosis
Sialidosis is one of the Oligosaccharide family of Lysosomal Storage Diseases. The International Society for Mannosidosis & Related Diseases is proud to present the story of Alexander Skojec and his family’s fight, through the intervention of his father, to raise awareness for this very rare disorder.


Trajano
Hello, my name is Trajano Cerna. I am from Quito-Ecuador, and I live in the United States. I went to School of Medicine for 2 years in Ecuador. I enjoy walking and doing exercises in the park. And I like drinking natural juices. Read more about my journey here.




ISMRD works to connect families and raise awareness about the nine Glycoprotein Storage Diseases. Stories help to get the word out about the real life experiences of families within ISMRD. We are so happy you’ve found this form and are interested in sharing your story!
We ask for about 1-2 paragraphs explaining your or your family member’s journey journey, when diagnosed, favorite things (activities, foods, etc), and how you heard about ISMRD. We also ask for a few photos to include when we post the story. If you have any questions, email info@ismrd.org.